本文主要是介绍「BioNano系列」那些Bionano未覆盖的区域是什么?,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
在「Bionano系列」光学图谱混合组装应该怎么做?这篇文章中,我展示了下面这张图。
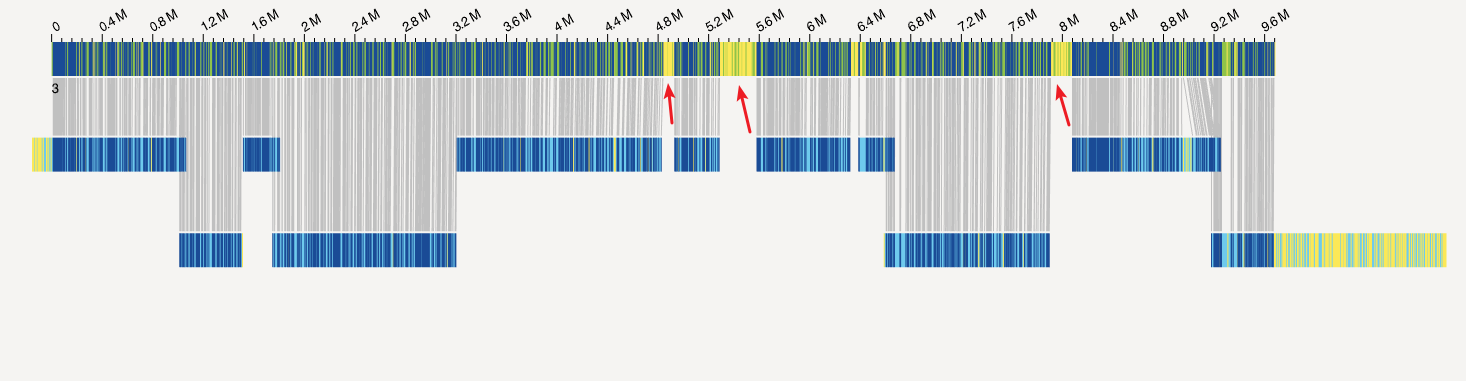
和之前的图不同的是,我加了几个箭头,这些箭头所指向的区域的特征就是,这些区域并未被Bionano所覆盖。如果不去思考这些区域到底是什么,直接进行混合组装,那么这其实对最后结果的不负责任。因为这完全可能是组装软件没有正确的处理错误的overlap,将不应该连接的序列连接在一起(尽管这个概率不高)。
我的直观猜测就是,这些区域应该是重复序列区域。毕竟Bionano标记技术依赖于酶识别特定位点进行酶切加上荧光标记,重复序列要么会因酶切密度太高,相机的分辨率达不到,而识别失败,要么是酶切位点过少,信号太弱。
那么我应该如何验证这个猜想?通过几天的文献翻阅和尝试,我用重复序列数量和基因数量的相对比值进行衡量。
命令行的代码如下(没有考虑文件的相对位置)
# 利用拟南芥的原本CDS进行注释
gmap_build -D index -d R05C0144 ../R05C0144.fa &
gmap -t 20 -D index -d R05C0144 -f gff3_gene ../Athaliana_cds.fa > cds_gene.gff3 2> log.txt &
# 重复序列注释
RepeatMasker -e ncbi -species arabidopsis -pa 30 -gff -dir . ../R05C0144.fa &
# GFF转成BED
awk 'BEGIN{OFS="\t"} {print $1,$4,$5}' ../repeat_annotation/R05C0144.fa.out.gff > repeat.bed
grep -w 'gene' ../gene_annotation/cds_gene.gff3| awk 'BEGIN{OFS="\t"} {print $1,$4,$5}' | bedtools sort -i - > gene.bed
# 统计
bedtools makewindows -w 100000 -g ../R05C0144.txt > windows_100k.bed
bedtools coverage -a windows_100k.bed -b repeat.bed > repeat_stat.bed
bedtools coverage -a windows_100k.bed -b gene.bed > gene_stat.bed
R代码如下
gene_df <- read.table("R05C0144/feature_stat/gene_stat.bed",sep="\t", stringsAsFactors = F)
repeat_df <- read.table("R05C0144/feature_stat/repeat_stat.bed",sep="\t", stringsAsFactors = F)options(scipen=999)
contig <- "contig2"repeat_ctg <- repeat_df[repeat_df$V1 == contig,]
gene_ctg <- gene_df[gene_df$V1 == contig,]combine_df <- data.frame(pos=(repeat_ctg$V2 + repeat_ctg$V3) / 2,repeat_num=repeat_ctg$V4,gene_num=gene_ctg$V4)
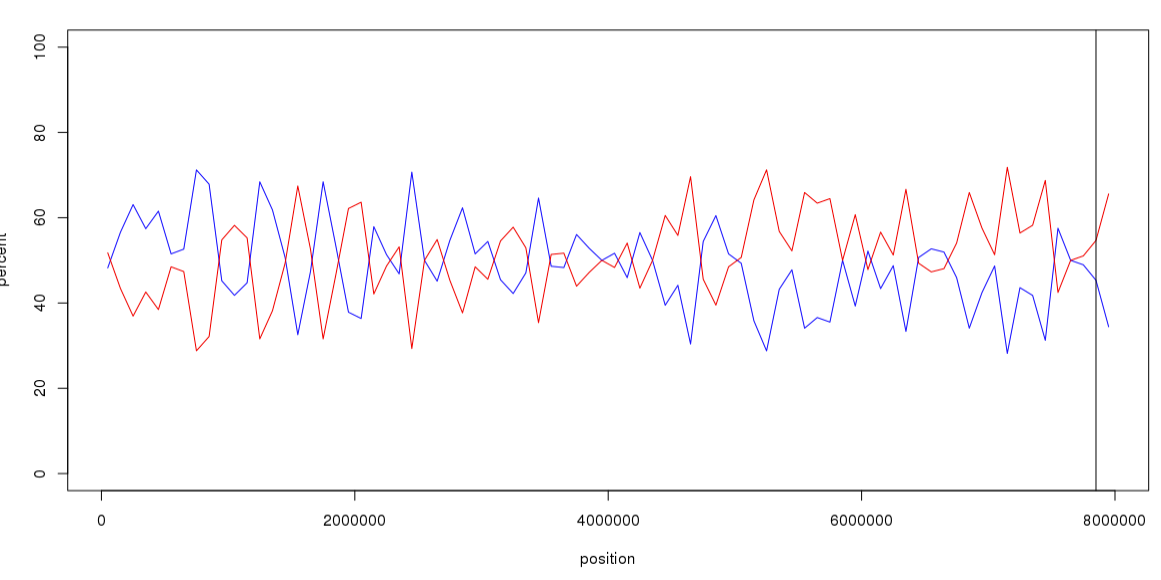
combine_df$total = combine_df$repeat_num + combine_df$gene_numcombine_df$gene_ratio <- combine_df$gene_num / combine_df$total * 100combine_df$repeat_ratio <- combine_df$repeat_num / combine_df$total * 100plot(combine_df$pos, combine_df$gene_ratio, type="l", ylim=c(0,100),xlab="position",ylab="percent",col="blue")
lines(combine_df$pos, combine_df$repeat_ratio, col="red")
abline(v=7.85*1e6)
我检查了一些区间,的确是重复序列比例高于基因比例,当然还有一些区间不是。说明重复序列并不是光学图谱未覆盖的主要原因。
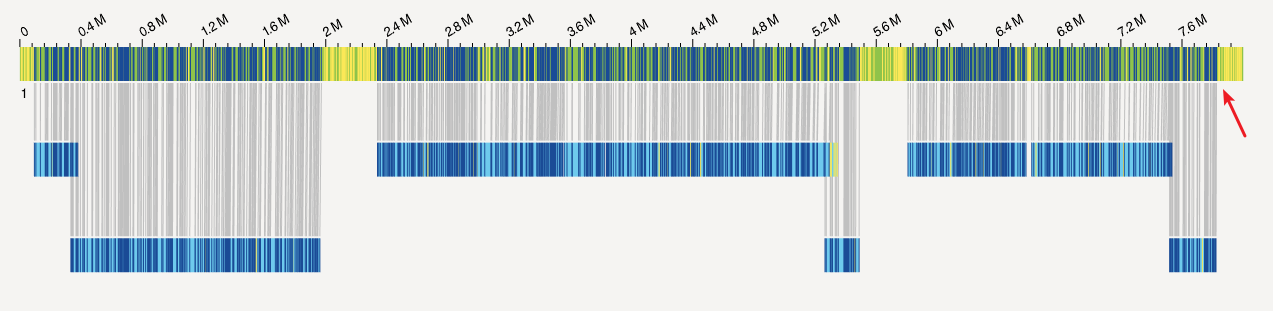
当然对于拟南芥这种有着高质量基因组的物种而言,我们还可以进行共线性分析。不过对于这些N50在4M左右,而且低杂合的基因组,其实都不需要太操心这种错误。
我这里也就验证了一种可能性,后续还得检查了一下其他原因,说不定仅仅是光学图谱的深度不够而已。
这篇关于「BioNano系列」那些Bionano未覆盖的区域是什么?的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






