本文主要是介绍WGDI之深入理解blockinfo输出结果,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
blockinfo模块输出文件以csv格式进行存放,共23列,可以用EXCEL直接打开。
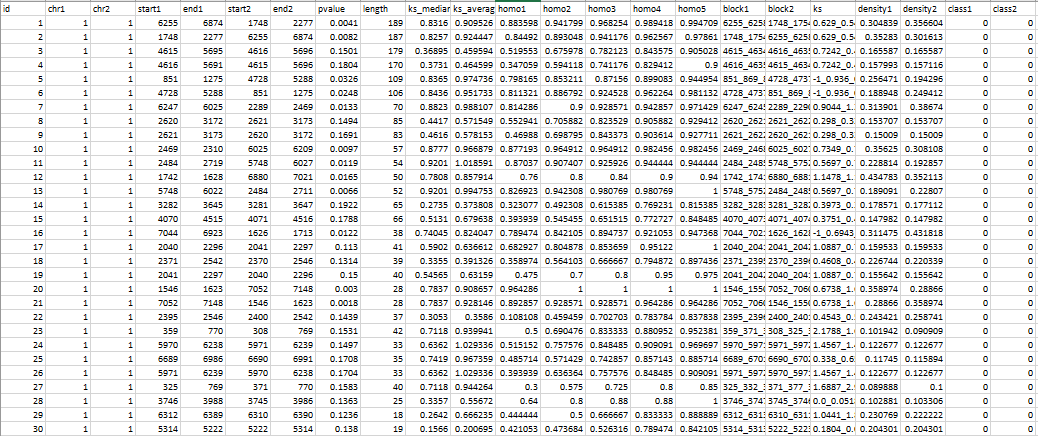
其中16列非常容易裂解,描述如下
id即共线性的结果的唯一标识chr1,start1,end1即参考基因组(点图的左边)的共线性范围(对应GFF1的位置)chr2,start2,end2即参考基因组(点图的上边)的共线性范围(对应GFF2的位置)pvalue即共线性结果评估,常常认为小于0.01的更合理些length即共线性片段中基因对数目ks_median即共线性片段上所有基因对ks的中位数(主要用来评判ks分布的)ks_average即共线性片段上所有基因对ks的平均值block1,block2分别为共线性片段上基因order的位置。ks共线性片段上所有基因对的ks值density1,density2共线性片段的基因分布密集程度。值越小表示稀疏。
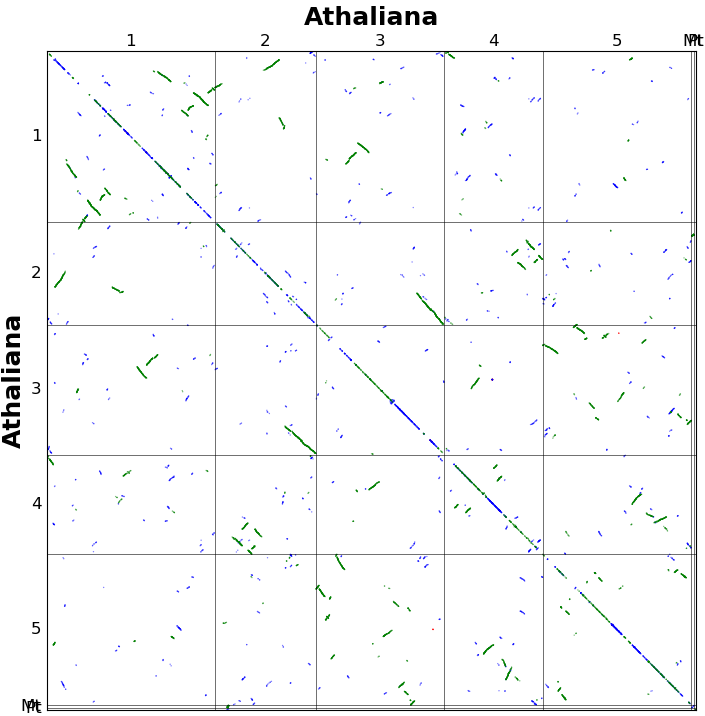
最后两列,class1和 class2会在 alignment 模块中用到,对应的是两个block分组,默认值是0表示两个block是同一组。这两列后期需要自己根据覆盖率,染色体核型等多个方面进行确定。举个例子,我们可以根据 homo1 的取值范围对class1进行赋值,例如-1~-0.5 是 1,-0.5 ~ 0.5 是2,0.5~1是3,最后在alignment中会就会用三种颜色来展示,例如下图的1,2,3分别对应red,blue,green.
中间的homo1,homo2,homo3,homo4,homo5并非那么直观,先说结论:
这里的homoN(N=1,2,3,4,5) 表示一个基因有N个最佳匹配时的取值
N由mutiple参数确定,对应点阵图(dotplot)中的红点
multiple的取值一般取1即可,表示最近一次的WGD可能是一次二倍化事件,因此每个基因只会有一个最佳匹配。如果设置为2,可能是一次3倍化,每个基因由两个最佳匹配。当然实际情况可能会更加复杂,比如说异源四倍体,或者异源六倍体,或者没有多倍化只是小规模的基因复制(small-scale gene duplication) 等情况,也会影响multiple的设置。
homoN会在后面过滤共线性区块时用到,一般最近的WGD事件所产生的共线性区块会比较接近1,而古老的WGD产生的共线性区块则接近-1.
接着,我们将根据源代码 blast_homo和blast_position 来说明结算过程。
首先需要用到blast_homo函数,用来输出每个基因对在不同最佳匹配情况下的取值(-1,0,1)。
def blast_homo(self, blast, gff1, gff2, repeat_number):index = [group.sort_values(by=11, ascending=False)[:repeat_number].index.tolist()for name, group in blast.groupby([0])]blast = blast.loc[np.concatenate(np.array([k[:repeat_number] for k in index], dtype=object)), [0, 1]]blast = blast.assign(homo1=np.nan, homo2=np.nan,homo3=np.nan, homo4=np.nan, homo5=np.nan)for i in range(1, 6):bluenum = i+5redindex = np.concatenate(np.array([k[:i] for k in index], dtype=object))blueindex = np.concatenate(np.array([k[i:bluenum] for k in index], dtype=object))grayindex = np.concatenate(np.array([k[bluenum:repeat_number] for k in index], dtype=object))blast.loc[redindex, 'homo'+str(i)] = 1blast.loc[blueindex, 'homo'+str(i)] = 0blast.loc[grayindex, 'homo'+str(i)] = -1return blastfor循环前的代码作用是提取每个基因BLAST后的前N个最佳结果。循环的作用基因对进行赋值,主要规则是基因对如果在点图中为红色,赋值为1,蓝色赋值为0,灰色赋值为-1。
homo1 对应 redindex = 0:1, bluenum = 1:6, grayindex = 6:repeat_number
homo2 对应redindex = 0:2, bluenum = 2:7, grayindex = 7:repeat_number
...
homo5对应redindex=0:5, bluenum=5:10, grayindex = 10:repeat_number
最终函数返回的就是每个基因对,在不同最佳匹配数下的赋值结果。
0 1 homo1 homo2 homo3 homo4 homo5
185893 AT1G01010 AT4G01550 1.0 1.0 1.0 1.0 1.0
185894 AT1G01010 AT1G02230 0.0 1.0 1.0 1.0 1.0
185899 AT1G01010 AT4G35580 -1.0 0.0 0.0 0.0 0.0
185900 AT1G01010 AT1G33060 -1.0 -1.0 0.0 0.0 0.0
185901 AT1G01010 AT3G49530 -1.0 -1.0 -1.0 0.0 0.0
185902 AT1G01010 AT5G24590 -1.0 -1.0 -1.0 -1.0 0.0
250822 AT1G01030 AT1G13260 0.0 0.0 0.0 1.0 1.0
250823 AT1G01030 AT1G68840 0.0 0.0 0.0 0.0 1.0
250825 AT1G01030 AT1G25560 0.0 0.0 0.0 0.0 0.0
250826 AT1G01030 AT3G25730 -1.0 0.0 0.0 0.0 0.0
250824 AT1G01030 AT5G06250 -1.0 -1.0 0.0 0.0 0.0然后block_position函数, 会用 for k in block[1]的循环提取每个共线性区块中每个基因对的homo值,然后用 df = pd.DataFrame(blk_homo) 和 homo = df.mean().values求均值。
def block_position(self, collinearity, blast, gff1, gff2, ks):data = []for block in collinearity:blk_homo, blk_ks = [], []if block[1][0][0] not in gff1.index or block[1][0][2] not in gff2.index:continuechr1, chr2 = gff1.loc[block[1][0][0],'chr'], gff2.loc[block[1][0][2], 'chr']array1, array2 = [float(i[1]) for i in block[1]], [float(i[3]) for i in block[1]]start1, end1 = array1[0], array1[-1]start2, end2 = array2[0], array2[-1]block1, block2 = [], []## 提取block中对应基因对的homo值for k in block[1]:block1.append(int(float(k[1])))block2.append(int(float(k[3])))if k[0]+","+k[2] in ks.index:pair_ks = ks[str(k[0])+","+str(k[2])]blk_ks.append(pair_ks)elif k[2]+","+k[0] in ks.index:pair_ks = ks[str(k[2])+","+str(k[0])]blk_ks.append(pair_ks)else:blk_ks.append(-1)if k[0]+","+k[2] not in blast.index:continueblk_homo.append(blast.loc[k[0]+","+k[2], ['homo'+str(i) for i in range(1, 6)]].values.tolist())ks_arr = [k for k in blk_ks if k >= 0]if len(ks_arr) == 0:ks_median = -1ks_average = -1else:arr_ks = [k for k in blk_ks if k >= 0]ks_median = base.get_median(arr_ks)ks_average = sum(arr_ks)/len(arr_ks)# 对5列homo值求均值 df = pd.DataFrame(blk_homo)homo = df.mean().valuesif len(homo) == 0:homo = [-1, -1, -1, -1, -1]blkks = '_'.join([str(k) for k in blk_ks])block1 = '_'.join([str(k) for k in block1])block2 = '_'.join([str(k) for k in block2])data.append([block[0], chr1, chr2, start1, end1, start2, end2, block[2], len(block[1]), ks_median, ks_average, homo[0], homo[1], homo[2], homo[3], homo[4], block1, block2, blkks])data = pd.DataFrame(data, columns=['id', 'chr1', 'chr2', 'start1', 'end1', 'start2', 'end2','pvalue', 'length', 'ks_median', 'ks_average', 'homo1', 'homo2', 'homo3','homo4', 'homo5', 'block1', 'block2', 'ks'])data['density1'] = data['length'] / \((data['end1']-data['start1']).abs()+1)data['density2'] = data['length'] / \((data['end2']-data['start2']).abs()+1)return data最终得到的homo1的homo5,是不同最佳匹配基因数下计算的值。如果共线性的点大部分为红色,那么该值接近于1;如果共线性的点大部分为蓝色,那么该值接近于0;如果共线性的点大部分为灰色,那么该值接近于-1。也就是我们可以根据最初的点图中的颜色来确定将来筛选不同WGD事件所产生共线性区块。
这也就是为什么homoN可以作为共线性片段的筛选标准。
这篇关于WGDI之深入理解blockinfo输出结果的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






