本文主要是介绍对snpeff注释的理解性问题3.0版本,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
需要做汇报PPT但是PPT不能呈现所有内容,先用文本记录一下。
在了解为什么我们要进行注释之前,我们得了解一下什么叫做snp
1.SNP是什么
不得不说一下我们的snp的来源,生物多样性:生态系统多样性,物种多样性,遗传多态性,其中DNA多态性,Polymorphism at the DNA level includes a wide range of variations from single base pair change, many base pairs, and repeated sequences.
因此,我们知道DNA序列变异的种类(DNA多态性的种类):
1.1 SNP(单个):Single Nucleotide Polymorphism(Commonly used to also include rare variants (SNVs))
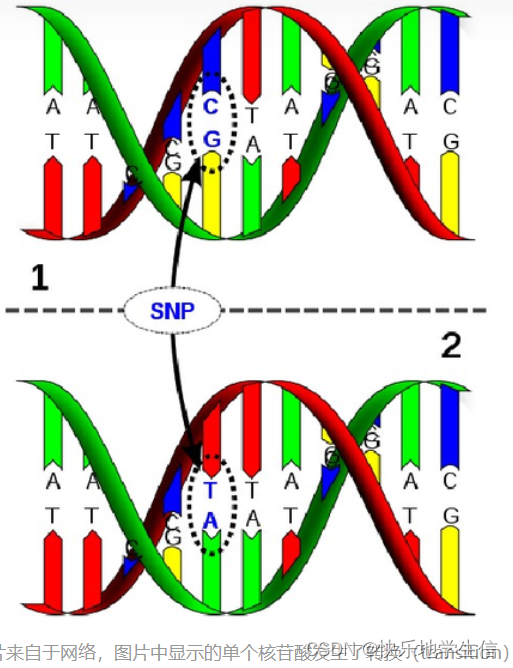
1.2 INDEL(小于50bp的小片段的插入缺失称为Indel):Indel is a molecular biology term for an insertion or deletion of bases in the genome of an organism. It is classified among small genetic variations, measuring from 1 to 10 000 base pairs in length...
1.3 VNTR(数目可变串联重复序列),
1.4 SSR(简单重复序列),
1.5 Rearrangement (应该是染色体层面的),
1.6 限制性片段长度多态性(RFLP)
在我查看资料总结的时候,看了的一些文章
单核苷酸多态性(SNP) - 知乎 (zhihu.com)
生信路漫漫 | 变异检测篇—InDel突变 - 知乎 (zhihu.com)
Science Bulletin | 一个Liddle综合征新SCNN1B基因移码突变的鉴定 (qq.com)
Autosomal Dominant Inheritance - an overview | ScienceDirect Topics
vcf文件中短序列插入缺失(INDEL)标准化 - 简书 (jianshu.com)
很好我们现在大概能理解SNP是什么意思了,现在我们来知道我们为什么要对SNP进行注释,在此之前我们得认识一下snpeff这个注释软件
2.SnpEff软件
这个是官网Running SnpEff - SnpEff & SnpSift (pcingola.github.io)
至于注释的方法,大家查看我写的另外一篇文章,snpeff注释最全.包括错误分析和理解.2.0版-CSDN博客
我们知道SnpEff构建基因库,其实需要两个文件,一个是gff文件,一个是fa文件,这个两个文件分别是什么,
gff文件:
基因组注释文件是包含GFF,GTF两种主要格式,用于高通量测序中对已经map到参考基因组的reads做注释。(基因组测序时需要先将基因组打断成DNA片段,然后再建库测序。reads指的是测序仪单次测序所得到的碱基序列)。至于其他了解可以去查看一下这篇文章,目前我没有用到,所以这里不做多的解释。
基因组注释文件(GFF,GTF)下载的四种方法 - 知乎 (zhihu.com)
1.GFF文件(General Feature Format):这是一种常见的基因组注释文件格式,它包含了基因组上的各种生物学特征的位置信息,如基因、外显子、内含子、转录本、蛋白质编码区域等。GFF文件将这些特征以层次结构的形式进行组织,并提供了它们在基因组上的起始位置、终止位置、方向等信息。
fa文件:
我们不得不说
生信分析必须了解的4种文件格式 - 知乎 (zhihu.com)
基因组注释文件(二)| gff 和 gtf文件格式说明 - 简书 (jianshu.com)
2.FA文件(FASTA格式):这是一种常见的DNA或蛋白质序列文件格式,每条序列通常以一个唯一的标识符开始,后面跟着该序列的碱基(DNA)或氨基酸(蛋白质)序列。FA文件提供了基因组的序列信息,用于将变异与基因组序列对应起来。
不管如何说,注释的理由就是(个人理解):从注释的过程中知道SNP具体信息,方便后续操作。
3.其他一些东西
之前我因为报错太多,主要是位点不在染色体范围内报错很多,7%左右,所以我们得验证一下这个是否真的是位点不在染色体范围内,
0.EXCEL表格的方式查找可能会更快,但是我们的数据太大了
1.galaxy的operate on genomic intervals 或者是UCSC的data interagete
本来还有一篇文章的,实在是没找到,下次我遇到了,我在加上去
高通量测序分析——从基因组重测序数据中获取突变信息 - 知乎 (zhihu.com)
Galaxy (usegalaxy.org)
2.敲代码,寻找这个范围
由于我技术不过关卡了很久,大家可以试试,我改天在尝试一下,下次写出来直接公示
3,最后我是怎么发现的,说实话这是一个玄学,
半夜两三点的时候,我用代码跑linux的时候,无意间看到最后一个是报错信息,也就是说,报错信息会直接体现出来,把注释好的vcf放在在kate里面,不知道说啥,亏我找了那么久,不然我真得通宵了。
4.其他注释的软件和一些相关文献
因为种种原因我还是没有完全使用其他的注释,但是这边我也想记录一下其他注释软件,上面有一篇文章有一些介绍,在这里我汇总一下:
1.SnpEff,
我们就不多说了,snpEFF是一个用于注释单核苷酸多态性(SNP)和小型插入/删除(Indel)的工具具。。它能够根据变异的位置、类型和影响等信息,提供关于这些变异可能的功能效果和相关注释的预测。例如非同义突变、错义突变和无义突变等,根据数据库提供的信息进行变异分类。意思就是,在寻找与疾病相关的遗传变异或解释个体基因组数据中的变异时,通过它可以帮助研究人员更好更快地理解变异对蛋白质结构和功能的影响,从而揭示潜在的影响。
snpEFF的优势在于它具有开放源代码、灵活性高、易于自定义和扩展等特点,
2.ANNOVAR
是由perl编写的程序,学术用途可以免费试用,一个常用的基因变异注释工具,它可以对基因组变异进行功能注释和分类。有机会试一下。
ANNOVAR | 变异注释【上】 - 简书 (jianshu.com)
ANNOVAR 注释|自建数据库 - 简书 (jianshu.com)
3.vep注释
是由Ensembl开发的一个广泛使用的基因变异注释工具,可以识别和注释各种类型的变异,如单核苷酸变异、插入/删除、结构变异等。VEP还提供了丰富的注释信息,包括变异的功能、频率、疾病相关性等,支持多个数据库源,网上有很多教程。
Tutorial (ensembl.org)
安装VEP及其注释数据库-腾讯云开发者社区-腾讯云 (tencent.com)
SNP注释神器——VEP(生信)_vep注释_小飞棍来喽~的博客-CSDN博客
VEP--强大的变异注释工具 | GeneDock 文档
用 VEP 注释突变数据-腾讯云开发者社区-腾讯云 (tencent.com)
大底上,conda安装,自建数据库还是需要gff文件,这个我粗略的搜寻一下,好像没有自建数据库的文章,也可能是我查错了,据说是自建不太灵活,过年回去试试。
4.bedtools
进行gwas基因注释,这可以直接去看邓飞老师的。使用bedtools进行gwas基因注释 - 知乎 (zhihu.com)
下次的学习注释的目标,
1,这几个注释都可以回家没事尝试一下。
2,Galaxy (usegalaxy.org)
3, UCSC的data interagete
当时看这个的时候就很懵,不太能理解,主要是一下软件的应用以及软件在注释上的一些不同之处。
最后,还说一点,我们得查看一下一些文献,虽然只能是粗略的检索一下
香菇L808细胞核与线粒体基因组注释分析 - 中国知网 (cnki.net)
基于高效SNP芯片的小麦产量相关性状全基因组关联分析 - 中国知网 (cnki.net)
对全球鸡种群的精细结构和混合的研究揭示了种群与育种史上重要事件之间的联系 - PMC (nih.gov)
实在是没啥好看的,我看得过于混乱, SnpEff注释和其他注释软件的一些文章
CooVar:共现变异分析仪 |BMC 研究简报 |全文 (biomedcentral.com)
Variant Classification: A comparison of Annovar, SNPeff and VEP (goldenhelix.com)
主要是我并不知道,这个注释的基因在我们整个文章中怎么使用, 缺乏检索能力。
这篇关于对snpeff注释的理解性问题3.0版本的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!








