本文主要是介绍PNAS | 蛋白质结构预测屈服于机器学习,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
今天为大家介绍的是来自James E. Rothman的一篇短文。今年的阿尔伯特·拉斯克基础医学研究奖表彰了AlphaFold的发明,这是蛋白质研究历史上的一项革命性进展,首次提供了凭借序列信息就能够准确预测绝大多数蛋白质的三维氨基酸排列的实际能力。这一非凡的成就是由Demis Hassabis、John Jumper以及他们在Google DeepMind和其他合作者的同事们共同取得的,它建立在几十年的实验性蛋白质结构确定(结构生物学)和多种融合生物启发的统计方法的渐进发展基础之上。但是,当Jumper和Hassabis将创新的基于神经网络的机器学习方法融入其中时,结果引起了轰动。实现半个世纪以来的蛋白质结构预测梦想已经加速了化学、生物学和医学等多个领域的进展和创新。

蛋白质
在1838年,荷兰化学家G. J. Mulder创造了一个科学术语——蛋白质,这个术语被选得再合适不过。它源自希腊词汇"prōteios",意味着"第一等级"。蛋白质在接下来的六十年中一直没有明确定义,直到逐渐(从1900年到1965年)揭示出所有蛋白质都是由20种基本氨基酸组成的多肽链,其序列由遗传密码规定,并且能够折叠成独特的结构(构象),这些结构决定了它们的化学性质,从而影响生物功能。毫无疑问,蛋白质是"第一等级"的,因为细胞内几乎没有不涉及它们的生物过程。
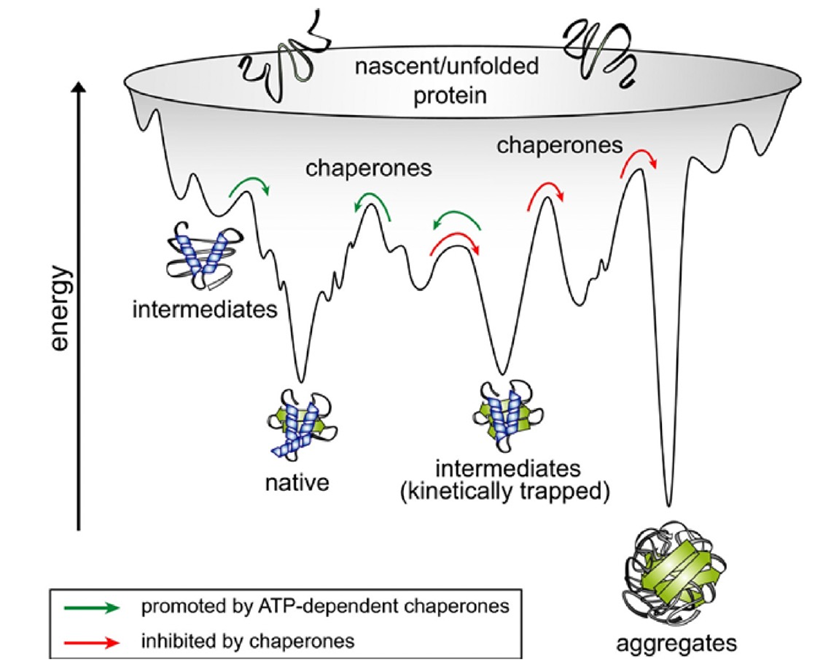
到了20世纪60年代,了解蛋白质如何折叠成独特的结构成为生物学的一个核心问题,人们期望能够预测蛋白质的结构将带来重大的益处。早期就已认识到,每个多肽链理论上可以假定有着天文数字级的可能构象,其中绝大多数会导致聚集和沉淀。然后在1961年,使用小型分泌酶核酸酶,Anfinsen证明了最简单的可能性成立——氨基酸序列本身可以决定链的折叠方式,而在这个过程中不需要额外的遗传信息。这一关键性实验将蛋白质折叠重新定义为物理问题,其简单思想是折叠状态是多肽链的最低可访问能量状态。但是,相应能量景观的极端复杂性,有许多局部能量井来捕获折叠中间体(图1),使得直接计算甚至模拟在除了最简单的情况以外几乎是棘手的,即使在计算能力呈指数增长的时期也是如此。
到了1990年,人们明白了生物细胞中的蛋白质折叠更加复杂,需要编码分子伴侣的基因。实际上,在高度进化的有机体中普遍存在的多域和多亚基蛋白质通常不会在Anfinsen风格的实验中高效或迅速地重新折叠。这绝不否定Anfinsen的核心思想,即最终的构象完全由多肽链的物理性质决定。但是,分子伴侣作为催化剂促进蛋白质折叠的发现是细胞生物学的一个基本进展。分子伴侣蛋白质本质上是ATP酶,通过将ATP水解的能量引导到折叠多肽链的能量陷阱中,"踢"出折叠多肽链,或者在首次避免这些陷阱(图1)中,同时屏蔽疏水侧链以防止聚集。通过这样做,分子伴侣蛋白质通过加速跨越能量景观的运动来催化折叠的整个过程。
结构预测
随着逐渐明显的趋势,直接的、仅基于物理的方法在没有像量子计算那样的革命性突破的情况下最终无法达到原子级的准确性,到了2010年左右,研究人员开始转向受生物启发的统计方法。其中一个基础性思想是,即使在氨基酸序列中它们彼此距离很远,但在折叠结构中物理上接触的氨基酸将以相关的方式在进化中变化。这些思想被纳入同源蛋白质的多序列比对(MSAs)中,这对所有预测算法都至关重要,包括AlphaFold。另一个基础性的统计概念涉及结构模板。除了同源序列,同源的三维结构也可以作为直接的蛋白质结构预测的起点。最初,基于模板的预测是主导方法,但协同进化方法在早期的2000年代,DNA序列的信息迅猛增长超越了蛋白质结构,于是占据了主导地位。在过去的10年中,随着冷冻电镜结构测定的出现,绕过了晶化的瓶颈,研究人员又开始关注蛋白质结构。
最重要的因素被证明是机器学习,它使得来自MSAs和模板以及其他数据的信息能够更加高效地被利用。尽管MSAs和模板的信息可以在单独的流程中使用,但它们也可以以交互方式同时使用。这基本上是像AlphaFold这样的"神经网络"系统在从数据中学习最佳的组合方式时所做的。为了客观评估进展,折叠研究领域建立了一个两年一次的比赛,即蛋白质结构预测的关键评估(CASP),该比赛测量了针对当时尚未公开的实验确定的结构的竞争方法的比较准确性。DeepMind团队在第13届会议中首次亮相,并且引起了广泛关注,他们的AlphaFold(版本1)在96个竞争对手中表现出色,但仍然远未达到原子级的预测准确性。他们的算法为43个测试蛋白质中的25个生成了最佳结构。
然而,Hassabis知道这还不够好。他们首先尝试将AlphaFold1推向极限。大约在CASP13后的六个月后,意识到它无法达到我们想要的原子级准确性,无法真正解决问题,也无法对实验学家和生物学家有所帮助。因此做出了我们需要重新开始的决定...但在那之后的大约六个月到一年内,情况变得更糟,而不是更好。AlphaFold2系统(早期版本)比AlphaFold1差得多。在准确性方面似乎退步是非常可怕的时期。John Jumper于2017年加入了DeepMind,担任研究科学家,他及时为CASP13/AlphaFold1项目提供了一些新的想法,这些想法后来成熟为AlphaFold2。Hassabis显然对Jumper印象深刻,并在2018年7月提拔他为AlphaFold2的负责人。Hassabis认识到Jumper在蛋白质物理学和机器学习领域的跨学科背景的重要性。至关重要的是,他们共享一个大胆的信念,即在AlphaFold2性能不断恶化的情况下,仍然可以解决预测问题(即准确性可以达到基因组尺度的1埃)。
最终,Jumper和Hassabis带领他们的团队进行了完全的系统重构,包括许多创新。描述这些方法的《自然》论文长达约60页。尽管这篇2021年8月的发表是一个重要的里程碑,但关键事件实际上发生在一年前,当所有参赛者提交了他们的结果给CASP14竞赛(2020年5月至7月)。当结果揭晓时(2020年12月),结构生物学界感到震惊。AlphaFold2远远超越了所有竞争对手,将预测提升到了原子级准确度,实现了飞跃。
为什么AlphaFold可行
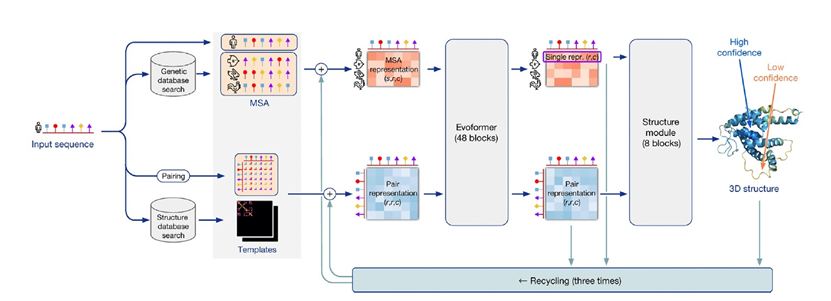
尽管AlphaFold神经网络是由人类设计的,就像人脑一样,我们可能永远无法完全理解它。AlphaFold是一种工程产品,其内部解剖结构是完全明确的(图2)。理解它究竟如何工作,就成了一个独立的科学问题。在神经科学中,经典的方法是消融,字面意思是去除大脑的一部分,或更微妙地切断连接,甚至更微妙的是在细胞层面引入光遗传损伤,或者在人类情况下观察与病变相关的功能缺陷。Jumper、Hassabis和他们的团队进行了系统性的计算消融研究,明确证明了各种不同的机制以一种在很大程度上合并到整体的方式起作用。
设计中的一个基本机制是机器学习的人工神经网络,它捕捉了长程和短程相互作用,与以前的算法不同,以前的算法通常只考虑了局部或成对的相互作用,部分通过考虑多肽链几何结构的基本特征来实现。迭代细化的使用类似于人脑的工作方式,是其另一个核心概念。其他关键特征包括一个新的架构,用于联合嵌入MSAs和成对特征;一个新的输出表示和相关的损失,可以实现准确的端到端预测;使用中间损失来实现迭代细化;以及涉及自我蒸馏的新的培训程序。该网络分为两个主要阶段(图2)。首先,网络的主干通过重复使用称为Evoformer的新型网络块来处理输入,最终表示经过处理的MSAs和残基对,并包含基于注意力的组件。消融研究表明,在Evoformer中形成了一个具体的结构假设,并不断更新。Jumper和Hassabis在他们的论文中解释说,Evoformer中的关键创新包括在MSA和对表示之间交换信息的新机制,这使得可以直接推断空间和进化关系。这些更新本质上创建了预测结构收敛到正确解的轨迹,并可以视为在明确的分子动力学或蒙特卡洛模拟中的“动作”。其次,主干后面是结构模块,它以每个残基的坐标形式生成3D结构。随着每次迭代,这些结构迅速发展并细化,形成了具有原子细节的高度准确的蛋白质结构。关键的创新包括分解链结构,以允许对结构的所有部分进行同时细化,从而可能提供一种避免地景上多个能量陷阱(图1)的机制。
该网络是从开放获取的蛋白质数据银行(PDB)中进行训练的,这实际上是几代结构生物学家的努力累积而成。PDB目前包含约200,000个独特的蛋白质结构和相关序列,主要来自X射线晶体学和最近的冷冻电镜技术。然后,训练好的网络被用来预测另一个重要的公共库(Uniclust30)中约350,000个不同序列的结构。最后,使用混合PDB数据和预测结构数据集作为训练数据,从头开始训练了新的AlphaFold网络。这种自我蒸馏过程鼓励网络利用未伴随结构的序列数据,并观察到总体准确性的提高。
从GO开始
Hassabis和Jumper的成功展示了当两个领域融合时释放出的创造性能量。Demis Hassabis出生在英国伦敦,有希腊塞浦路斯和新加坡华人的血统,他是一个官方认可的神童,13岁时已经是公认的国际象棋大师,可以与成年人比赛。17岁时,他加入了Bullfrog Productions公司,在那里设计了畅销游戏《主题公园》等游戏。然后,他进入剑桥大学,在那里学习计算机科学和数学,并从生物学专业的朋友口中第一次听说了蛋白质折叠问题。毕业后(1997年),他加入了Lionhead Studios,然后创办了自己成功的公司Elixir Studios。"我一直在思考和致力于通用人工智能,甚至在大学时也是如此。我倾向于记下我认为有朝一日可能会变得可行的科学问题...而蛋白质折叠问题正是其中之一。"然而,他意识到他仍然需要学习如何专业地处理科学问题,因此他在伦敦大学学院学习了认知科学。在此之后,Hassabis创立了DeepMind(2014年被谷歌收购),他和同事最初专注于创建用于掌握游戏的学习算法,最终在2016年DeepMind的AlphaGo惊人地击败了传奇围棋选手李世石。"我们几乎是在从首尔AlphaGo比赛回来的那天开始的...我们从游戏开始,因为开发AI和测试各种东西更加高效...但最终,那从来不是最终目标。"在剑桥大学毕业近20年后,Hassabis准备翻出他的旧笔记。
John Jumper出生在阿肯色州。他的非传统之旅相对传统的从物理和数学(范德堡大学,2007年学士学位)开始,然后获得了前往剑桥大学的马歇尔奖学金。然而,他决定在金融领域担任“量化分析员”的职位,2008年申请加入了DE Shaw公司,这家由计算机科学家David Shaw创立的著名公司。碰巧的是,但直到他申请时,Jumper并不知道,Shaw已经从投资回归到基础研究,并专门设计了用于蛋白质折叠和药物发现的计算机设备,在D.E. Shaw Research工作,他被邀请加入Shaw的研究团队。在前沿计算领域工作了3年后,Jumper既致力于解决问题,也充分意识到他可能再也不会有如此强大的计算能力。听说DeepMind在围棋之后涉足蛋白质折叠领域,他设法将自己的简历送到Hassabis那里,而今年的阿尔伯特·拉斯克基础医学研究奖的获得者双人组就是在2017年开始合作的。
开普勒和牛顿不同
尽管我们为首次能够从基因序列准确预测基因组规模的蛋白质结构的发明而欢呼,但许多科学家也感到有些不安,因为我们并没有像预期的那样提取出可推广的传统科学解释原则。例如,基于开普勒的定律,我们可以准确预测行星的轨道,但开普勒的技术的全部威力只有在牛顿从中提取了古典力学原理后才被充分释放出来。有许多类似的例子。兰登的X射线,于1895年发现,并于1901年获得首个诺贝尔物理学奖,长时间以来在医学上产生了巨大的影响,尽管在最终从量子力学中理解它们之前,它们仍然是未解之谜。随着时间的推移,我们可以希望会出现有关蛋白质实际如何折叠(与最终形状不同)的类似深刻的见解和新原则。
未来的影响
毫无疑问,当前版本的AlphaFold只是一个开始。它已经在不断改进,以包括多亚单位蛋白质复合体。与任何革命性和强大的技术概念一样,它将吸引开放式创新,并以无法预测的方式发展,触及生物科学的各个角落。作者所知道的每个实验室都在使用AlphaFold并且是高度富有想象力的方式。即使在这些无数的探索远征中不可避免地会失败,也会在许多方面催化改进。然而,AlphaFold可能最长期的影响之一是预示AI即将成为生物学中被接受的、可靠的和有用的发现方法。今天的科学家大多接受了使用模型的培训,但并不真正信任它们,特别是如果他们无法明确理解这些模型。我们准备好信任模型并进行验证吗?
参考资料
Rothman, J. E. (2023). Starting at Go: Protein structure prediction succumbs to machine learning. Proceedings of the National Academy of Sciences, 120(39), e2311128120.
这篇关于PNAS | 蛋白质结构预测屈服于机器学习的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







