本文主要是介绍学习生物信息学的十条准则,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
介绍
测序技术正变得比以往任何时候都更加先进和实惠。作为回应,不断壮大的国际联盟,例如地球生物基因组计划 (EBP) 、万种脊椎动物基因组计划 (G10K)、全球无脊椎动物基因组联盟 (GIGA) 、5000种昆虫基因组(i5K)、万种植物基因组计划(10KP),还有许多组织制定了对地球上所有生命进行测序的宏伟计划。
这些联盟旨在利用基因组数据来揭示我们星球生物多样性的生物学秘密,并将这些知识应用于现实世界的问题,例如提高我们对物种进化的理解,协助保护受威胁的物种,以及确定新的医学目标,农业或工业用途。所有这些目标都依赖于有人来分析和理解海量的生物数据,这使得bioinformaticians比以往任何时候都更加抢手。许多具有生物学和遗传学背景的研究人员正在加紧迎接大数据分析的挑战,但是如果没有强大的计算和/或计算机科学。最近的一篇“十大简单规则”文章强调了生物信息学研究支持的重要性。
在这里,为有兴趣进入生物信息学领域以及那些开始生物信息学之旅的人提供 10 条简单规则。无论你是学生、经验丰富的生物学家或遗传学家,还是可能对这一新兴领域感兴趣的任何人。这些规则用于开始使用命令行学习生物信息学。
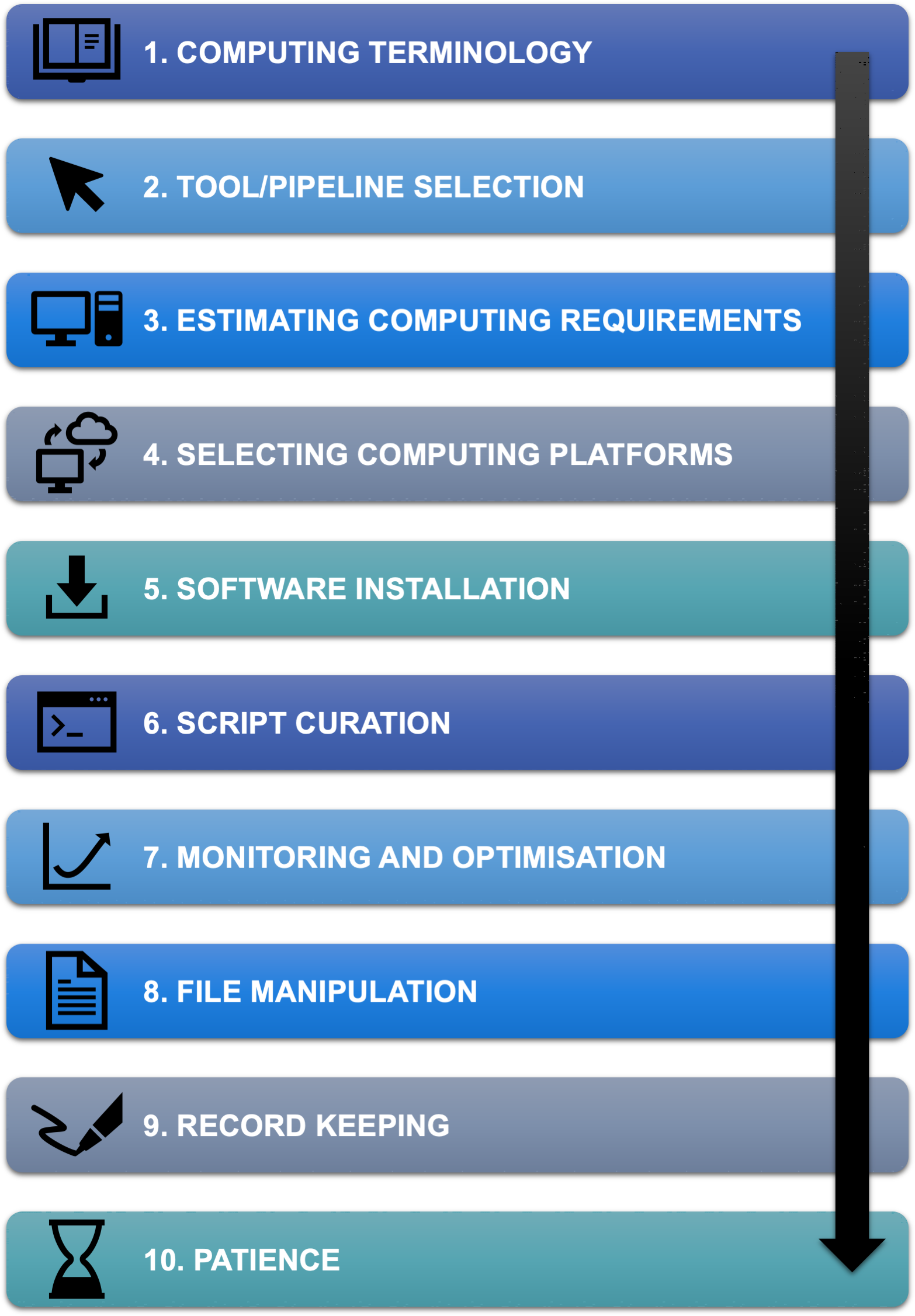
图 1.开始使用命令行生物信息学的 10 步过程
一、熟悉计算机术语
由于大量新术语,生物信息学之旅的第一步可能会让人不知所措。对计算和相关术语有基本的了解对于确定如何有效地运行生物信息学Pipeline非常有用。它还可以帮助你解决过程中的许多错误。
常见计算机术语的一些简单定义
- 算法
由计算机程序执行的一组规则或计算。某些算法可能更适合特定的数据集,并且可能在性能(例如,速度或准确性)方面存在差异。
- 中央处理单元 (CPU)
在计算节点或 VM 上执行实际计算的芯片。
- 计算节点
包含多个 CPU 和关联 RAM 的单个计算机。
- 核心
CPU 的一部分。单核处理器每个 CPU 包含 1 个内核,这意味着 CPU 和内核通常是可互换的术语。
- CPU time
CPU 实际处理数据所花费的时间(通常是 CPU 时间 ~ = Walltime * CPU 数)。
- 依赖项
成功执行的另一个工具或管道所需的软件。
- 可执行文件
包含工具/程序的文件。某些软件具有单个可执行文件,而其他软件具有针对不同命令/步骤的多个可执行文件。
- 高性能计算机 (HPC)
连接的计算节点的集合。
- 操作系统 (OS)
支持计算机基本功能的基础软件。一些最常见的基于 linux 的操作系统包括 Debian 发行版 (Ubuntu) 和 RedHat 发行版(Fedora 和 CentOS)的操作系统。
- 管道(Pipeline)
管道是由各种步骤(命令)和/或工具组成的工作流,它们处理给定的输入集以创建所需的输出文件。
- 编程语言
用于指示计算机执行特定任务的特定语法和规则。生物信息学中常用的编程语言包括 Bash、Python、Perl、R、C 和 C++。
- 随机存取存储器 (RAM)
临时存储 CPU 需要的所有信息(可以被关联节点或 VM 上的所有 CPU 访问)。
- 调度程序
管理在共享 HPC 环境中运行的作业(脚本)。一些常见的调度程序包括 SLURM、PBS、Torque 和 SGE。
- 脚本
包含要以单一编程语言执行的代码的文件。
- 线程
程序可以并发执行的计算数量——取决于内核数(通常 1 个内核 = 1 个线程)。
- 工具
对输入数据集执行分析以提取有意义的输出/信息的软件程序——工具、软件和程序通常可互换使用,但指的是生物信息学管道的核心组件。
- VM
虚拟机——类似于计算节点,因为它表现为一台计算机,包含所需数量的 CPU 和关联的 RAM(通常与云计算相关联)。
- Walltime
程序在我们的挂钟时间中运行所需的时间。
二、了解数据和需求,以确定要使用的工具或管道
这通常是最困难的步骤之一,因为对于每个特定的生物信息学分析,通常有许多不同的工具和管道可供选择。虽然可能会考虑创建自己的工具来执行特定任务,但通常情况下,已经有一个预先存在的工具可以满足需求,或者可能只需要稍作调整即可获得所需的结果。
清楚地了解你的数据和你想要提出的问题类型将大大有助于你选择工具或管道。选择最合适的管道或工具将取决于许多因素,包括:
- 目标物种和数据质量
根据特定物种的独特特征(例如,基因组大小、重复复杂性、倍性等)或基于数据质量(例如,scaffold长度、短reads与长reads、等等), 阅读有关类似物种的其他已发表论文将有助于对此进行定义。
- 可用计算资源和时间限制
某些软件可能基于不同的算法,这可能会导致计算资源和等待时间的显着减少或增加。某些共享的 HPC 基础设施可能存在时间限制,或者 RAM 或内核的数量可能是使用个人计算资源时的限制因素。在开始之前,了解个人计算或 HPC 基础设施的限制。
- 哪些工具是现成的
许多生物信息学管道和工具可供研究人员免费使用,但有些需要购买许可证。此外,某些工具/管道可能已经在你所需的计算基础设施上或通过当地的机构可用。有许多“标准”生物信息学命令行工具在各种基因组环境中具有广泛的适用性,因此很可能已经安装在共享基础设施上。此类示例包括 tabix、FastQC、samtools、vcftools/bcftools、bedtools、GATK、BWA、PLINK 和 BUSCO。此外,合作者或其他研究人员可能已经在特定基础设施上测试和优化了特定管道,因此已经为你克服了第一个障碍。
与从事类似项目的朋友交谈并通读文献通常是决定使用哪种软件进行特定分析的最佳方式。
有许多出版物对不同工具进行了基准测试,并比较了类似管道的优缺点。还有许多在线网络论坛(例如,BioStars) 这也可能有助于你的决策过程。
一定要搜索不同的网络论坛,看看其他研究人员是否也提出了与你相同或相似的问题(这种情况经常发生)。如果你找不到解决方案,请确保你发布的任何问题都清晰详细,并提供代码或错误示例,以便获得有用的回复和答案。
如果新的或不同的Pipleline看起来更适合你的数据或期望的结果,请不要害怕尝试。
三、估计你的计算需求
一旦你选择了所需的工具或管道,下一个关键步骤就是为你选择的分析估计所需的计算要求。估计你的需求不仅可以让你确定哪些平台可能最适合运行你的管道(例如,云还是 HPC,而且还可以减少对基本资源错误进行故障排除所花费的时间(例如,RAM 或储存空间不足)。
此外,在任何给定的计算基础设施上运行任何工具或管道之前,你需要确保有足够的可用磁盘空间。同样,对于云计算,你需要决定机器大小(核心和 RAM)以及分析所需的附加存储量。
默认情况下,许多生物信息学工具可以在单个内核上运行,但这会导致更长时间(通常受限于共享 HPC 基础架构)。增加内核数量可以大大减少你的挂机时间,尽管这与其他重要因素(例如 RAM 使用率、成本、排队时间等)之间通常存在平衡。
为你以前从未运行过的管道或从未测试过管道的物种估算计算需求可能有点棘手。
不过不要害怕,因为你可以在许多地方找到有关计算要求的信息。
首先,阅读你正在运行的管道/工具的文档。一些工具文档将提供所需计算资源的示例或提供建议。此外,许多程序将提供测试数据集以确保管道在使用你自己的数据集之前正常工作。如果工具文档未提供计算要求指南或示例数据集,你可能使用自己数据的较小子集进行初始测试。
文献还可以为已用于类似物种或样本大小的特定工具或管道的一般计算要求提供指南。有许多出版物将常见的生物信息学管道相互比较,以评估各种生物体的性能和结果。这些可以通过简单的引文搜索找到。最后,用于估算计算需求的另一个重要资源来自其他研究人员。与你所在领域中可能使用类似数据的其他人交谈或使用在线论坛(如 BioStars)将有助于了解所需的资源。
一般来说,32 个内核和 128 GB 的 RAM 通常足以让大多数常见的生物信息学管道在合理的时间范围内运行。话虽如此,某些程序可能需要的比这少得多,而其他程序可能需要更高的内存要求或实现更大的并行化。
四、探索不同的计算选项
在估计所选管道的计算要求后,你需要确定此类资源可用的位置以及最适合你需求的基础设施。
一些工具可能很容易在个人计算机上运行,但许多大型生物信息学管道(尤其是在处理具有大型基因组的生物体,如哺乳动物和植物时)需要远远超过标准 PC 的计算资源。许多机构拥有本地 HPC 或访问国家/国际 HPC。然而,前所未有的测序数据已经开始将这些共享基础设施推向极限。这些资源并不总是非常适合生物信息学管道的要求,例如它们的高 I/O 需求和“突发”性质。这就是为什么云计算越来越受到bioinformaticians的欢迎。
与传统的共享 HPC 资源相比,云计算提供了许多关键优势,包括:
能够为你希望使用的每个生物信息学工具或管道定制计算资源;
完全控制你的计算环境(即操作系统、软件安装、文件系统结构等);
没有排队系统导致更快的研究时间;
无限的可扩展性和易于再现性。
利用云资源还可以防止研究人员购买和维护自己的物理计算机硬件(这可能既耗时又昂贵,而且远不及可扩展性)。
总体而言,决定在何处运行分析将取决于你的数据/物种、你最容易访问的平台、你之前的经验、你的时间和你的预算。探索不同的计算选项将使你能够选择最适合你需求的基础架构,并使你能够适应快速发展的生物信息学世界。
五、了解软件安装的基础知识
当想要为你的生物信息学管道使用个人资源时,例如云 VM 或个人计算机,你需要熟悉所需工具的各种安装方法。虽然有时软件安装是作为某些共享 HPC 平台的服务提供的,但了解软件安装的基础知识有助于帮助你解决任何基于安装的错误并确定你可能自己在本地安装哪些软件(即,不需要 root 用户权限) )。安装软件的方法有很多种,但我们提供了 4 种主要方法,应该涵盖大多数生物信息学软件。
生物信息学工具的常用软件安装方法
包管理器
APT(高级软件包工具) 是一个软件包管理器,通常默认情况下已经安装在许多 Debian 发行版上,并且启用非常简单安装可用工具。APT 与各种核心库协同工作,以自动下载、配置和安装软件包及其依赖项。
通过 APT 可以获得许多常见的生物信息学工具,包括 NCBIblast+、samtools、hmmer、vcftools、bcftools、bedtools 等。如果在 RedHat 操作系统上工作,包管理器 YUM (Yellowdog Updater, Modified) 相当于 APT。
Conda
Conda 也是一个包管理工具,尽管它介于 APT 和 Containers 等包管理器之间(见下文),因为它也能够管理环境(即,软件集)。此功能使 conda 非常有用,特别是对于生物信息学软件而言,不同的管道可能使用相同的工具但需要特定工具的不同版本。Conda 允许你在自己的独立环境中轻松安装和运行管道,这样它们就不会相互干扰,并且还使你能够在新版本可用时轻松更新软件。
Bioconda 是 conda 的channel,专门研究生物信息学软件,包括无数最常用的生物信息学工具。此外,conda 还支持安装和管理流行的编程语言,如 python 或 R,以及它们各自的库和包。它是各级bioinformaticians的重要资源,特别有用。
Containers
容器将软件和所有依赖项,以及所有基础系统工具和系统库打包到一个单独的环境中,以便它们可以在不同的计算平台上可靠地运行。 Containers 类似于 conda 环境,但它们的不同之处在于容器绝对包含容器本身所需的一切(甚至包括基本操作系统)。有时更容易将容器视为安装一个单独的机器,它只使用与安装它的本地机器相同的计算资源和硬件。容器相对于 conda 环境的主要优势是易于重现,因为每次你想要运行或重新运行特定管道或使用特定工具时都可以拉出特定容器,无论你使用什么计算平台。conda 环境也可以实现可重复性,但这通常需要导出并跟踪保存的环境。
想要使用容器时有两个主要选项:Docker 或 Singularity
Docker 是最标准的容器服务,可从 DockerHub 或其他容器注册中心 (如 quay.io )获得数千个容器。
通过 bioconda 提供的生物信息学软件在 quay.io 上也有一个通过 BioContainers 架构的相应 docker 容器。这意味着许多常见的生物信息学软件和管道已经在容器化环境中可用。否则,一些软件开发人员会提供他们自己的容器,例如 Trinity(用于 RNA-seq 组装,详见)或 BUSCO v4(用于评估组装完整性,详见)。在一系列在线容器注册表中还有数千个其他公共 Docker 容器,它们可能包含你正在寻找的软件,或者始终可以选择为可重现的管道创建自己的 Docker 容器。
显然,Docker 可用于下载和使用 Docker 容器,但 Singularity 是另一个也可用于下载和使用 Docker 容器的程序(特别是在 HPC 环境中)。两者都有优点和缺点,因此通常取决于用户的喜好来选择哪个。如果你不熟悉容器,我建议你从 Singularity 开始。这不仅使你能够轻松地将容器化管道扩展到 HPC 环境,而且使从本地计算机向容器读取文件和从容器写入文件变得更加简单。
手动安装
如果上述方法均不适用于你选择的软件,你可能需要手动安装。此过程通常在软件文档中逐步解释,但通常涉及许多步骤,包括:
- 从 GitHub ( https://github.com)(或其他网站);
- 对源代码进行解包以提取其内容;
- 配置软件以检查你的环境并确保所有必需的依赖项都可用;
- 从源代码构建完成的软件;
- 安装软件,即将软件可执行文件、库和文档复制到所需位置。
这个过程是包管理器和容器自动为你做的。手动安装通常需要许多标准依赖项(例如 build-essential 包、dh-autoreconf 包和 libarchive-dev 包),因此在尝试手动安装之前使用 APT 安装这些通常很方便安装任何其他软件。你将收到在安装过程中可能丢失的任何其他必需依赖项的通知。
安装软件后,最好尝试使用 help 命令行选项(即 -h/—help/-help)或不使用任何参数运行该程序,以确保它已正确安装。
如果帮助选项显示有关运行程序和不同命令行选项的一些信息,通常表明你的软件已成功安装并准备就绪。
如果你的工具似乎不起作用,你可能需要确保你的工具的可执行文件(有时是其所需的依赖项)在你的路径中可用。
但是你的路径究竟是什么,为什么它很重要?
每当我们在命令中调用特定的输入文件或输出目录时,我们经常使用绝对路径或相对路径来显示该文件或目录在文件系统层次结构中的位置。我们也可以以相同的方式调用工具或可执行文件,尽管每次需要使用工具时都提供指向工具的路径效率不高。path 环境变量通过提供包含你可能希望执行的工具/可执行文件的目录列表来克服这个问题。
默认情况下,路径变量始终设置为包括一些标准目录,这些目录包括各种系统命令行实用程序。因此,为了确保可以在不指定程序路径的情况下在任何地方调用新程序,你可以将工具/可执行文件移动或复制到路径变量中已列出的目录中,或者将新目录添加到路径中包含程序的变量。
新目录可以临时(通过简单地导出包含添加目录的路径变量)或永久(通过编辑 your.bash_profile)添加到你的路径中。
另一件要注意的事情是,路径中目录的顺序很重要,因为如果在 2 个不同的目录中找到相同的程序(或具有相同名称的可执行文件),系统将使用路径中第一个找到的程序。在将新目录添加到你的路径要确定它们应位于路径列表中的位置时,请始终牢记这一点。[仅在这条规则中提到“路径”这个词的次数就应该强调路径的真正重要性——尽管在本文的其余部分不再提及它]。
六、仔细检查和测试你的脚本
换句话说,始终仔细检查(或三重检查)你的脚本并在此过程中的每一步执行测试运行。在运行管道之前,务必先通读软件文档,以确保你了解不同的输入、输出和分析选项。确保文档适用于正确版本的软件,因为特定的命令行选项可能会更改版本。许多生物信息学程序都有大量的在线文档,无论是通过他们的 GitHub 还是其他网站。大多数工具的基本文档都可以使用命令行帮助选项访问(这也是确定所需工具是否可用并正确安装的好方法。有时可以在源代码目录中的 README 文件中找到更详细的信息。
获得最终脚本后,必须对其进行快速测试,以确定是否存在任何会阻止脚本成功运行的即时错误。从导致找不到文件或目录或命令与无效选项混淆的简单拼写错误或语法错误,到无法找到所需的软件或软件配置不正确,依赖关系有问题。
七、监控和优化你的管道
一旦你的脚本运行起来,就必须监控你的管道以确定它是否有效地利用了你分配给它的计算资源。了解你的管道使用哪些资源可以帮助你扩展或缩减计算量,这样你就不会浪费资源或达到可能会减慢管道速度的资源限制。在共享 HPC 基础设施上,你通常可以从作业日志文件或特定于调度程序的命令中看到所使用的计算资源的摘要。诸如最大 RAM 和 CPU 使用率以及 CPU 时间和挂墙时间等指标可用于调整未来的脚本,以便它们请求所需的最佳资源量。这使管道能够高效运行,而无需任何不必要的排队时间。
在云中运行管道时可以进行更具体的监控,因为你可以完全控制所有计算资源。像 htop这样的简单程序可用于快速实时监控 CPU 和 RAM 使用率等基本指标,而更深入的程序如 Netdata 可以帮助使用数百个预配置的交互式图形实时和跨整个管道跟踪各种指标。
许多生物信息学管道本质上是“突发的”,这意味着单个管道中的不同步骤可能具有截然不同的计算要求。某些步骤/工具可能具有较高的内存要求,但仅使用少量内核,而其他步骤/工具可能会在大量内核上实现良好的多线程,但只需要最少的内存。了解每个步骤所需的计算资源可以帮助你分解管道并在不同的机器类型上运行每个阶段,以提高成本效率。监控整个管道的磁盘空间需求也很重要,因为许多生物信息学工具需要大量的临时存储空间,这些存储空间通常在管道完成后进行清理。附加存储在云中可能非常昂贵,因此确保你只请求必要的内容也将降低管道成本。
总体而言,生物信息学管道的监控是提高管道效率、优化计算资源、减少排队时间浪费和降低云成本的关键。
八、熟悉基本的 bash 命令
作为bioinformaticians,你的主要职责是理解生物数据集,这通常意味着操作、排序和过滤各种生物信息工具和管道的输入和输出文件。例如,你可能想要提取某个样本或某个感兴趣的基因的信息。或者在包含数据表的文件中,你可能希望按特定列对输出文件进行排序或选择包含特定值的行。你可能希望用列表中的相应名称替换某个 ID,或者对列中的值执行计算。
幸运的是,生物信息学中使用的许多输入和输出文件都是常规文本文件,因此可以轻松完成这些任务。人们可能会考虑使用常见的电子表格应用程序(例如 Microsoft excel)来执行这些任务;然而,虽然这对于小文件可能已经足够了,但 excel 不太喜欢有时数百万行数据,这些数据是许多常见生物信息学文件的特征。这是一些标准的 unix shell 命令行实用程序发挥作用的地方,即 grep、AWK 和 sed 实用程序。
全局正则表达式打印 (grep) 是一个命令行实用程序,它在文本文件中搜索正则表达式(即文本模式)并返回包含匹配表达式的行(表 1)。当想要根据特定单词或文本模式(例如,样本名称或基因组位置等)的存在对文件进行过滤或子集化时,此工具非常有用。
AWK是更广泛的命令行实用程序这使得能够基于列的文件更具体的文件操作(表1)。例如,AWK 可以返回列包含特定值或正则表达式的行;此外,它可以只输出特定的列,对列内的值执行计算,并一次处理多个文件。AWK 的广泛功能太强大了,无法在这里涵盖,但只要知道这个聪明的小工具可能会在任何bioinformaticians的心中占有特殊的位置。
Sed 具有基本的“查找和替换”用法,允许你转换文本中定义的模式。在最基本的形式中,sed 可以用另一个给定的单词替换一个单词,但也可以执行更有用的功能,例如删除某个模式之前或之后的所有内容或在文件中的某些位置添加文本。
当然,grep、AWK 和 sed 都有其局限性,更广泛的文件操作可能更适合 python 或 perl 脚本; 但对于生物信息学文件的简单处理、过滤和操作,只需看看这 3 个有用的命令行实用程序即可。
九、写下来!
之前的一篇“十条简单规则”文章强调了为bioinformaticians保留实验室笔记本的重要性,另一篇文章介绍了围绕科学软件文档的一些最佳实践。
这些文章中的许多组件都适用于我们在开始使用命令行生物信息学时将其写下来并保留有用笔记的规则。
可以在一组生物数据上运行的管道或分析的数量有时可能非常广泛,并且通常与不同参数、计算资源和/或工具的大量试验和错误相吻合。即使是那些记忆力很好的人,也会经常在发表时回顾结果并思考“我们为什么使用该工具?”,或者“我们最终决定为该分析选择哪些参数?”。保留详细的笔记可以成为真正的救星。不仅跟踪不同的脚本文件以及每个脚本所需的计算资源很重要,以及关于你选择特定工具的原因以及成功运行管道所必须进行的任何故障排除的随附说明。一个包含所有你最喜欢的命令和漂亮代码的易于访问的文档,这些代码可能会一次又一次地派上用场,也是必须的!熟悉有用的代码文本编辑器,如 VScode 或 Atom,以及花一些时间学习有用的标记语言(如 Markdown)将有助于保持详细、有条理和格式良好你正在使用的管道的脚本和文档。
如何保留笔记完全取决于你,但只需确保将所有内容组织得井井有条,保持最新状态并进行备份。此外,将你的脚本发布为补充材料中的降价文件可确保你的工作的实用性(和可引用性)。
十、耐心是关键
成为一名成功的bioinformaticians的第一个关键(我们一直保存到最后)是耐心。
你的大部分时间将用于解决软件安装、计算错误、管道错误、脚本错误或奇怪的结果。有些问题很容易解决,而有些问题可能需要相当长的时间。你可能会觉得,每向前迈出一步,都需要跨越另一个障碍。然而,如果你有耐心并克服遇到的每一个错误,那么征服生物信息学管道并将大量数字数据或 As、Ts、Cs 和 Gs 转化为具有生物学意义的东西的欣喜是值得的。此外,正如该领域过去许多“十大简单规则”文章所阐述的那样,当你遇到困难时,请不要害怕举手寻求帮助。
大多数时候,在你之前的某个人曾处于完全相同的情况并遇到相同的错误或解决了类似的问题。当事情没有按计划进行时,Google 将成为你最好的朋友和第一站。在无休止的谷歌搜索无处可去的罕见情况下,与同行交谈并联系生物信息学社区;人们通常非常乐意分享他们的知识并测试他们解决问题的能力。
结论
在全基因组测序的新时代, bioinformaticians 比以往任何时候都更加抢手。进入命令行生物信息学的世界可能是一个陡峭的学习曲线,但却是一个值得承担的挑战。
希望以上十点内容能够让任何有抱负的生信人在解开隐藏在其生物数据集深处的有意义的影响的旅程中领先一步。
参考文献
Brandies P A, Hogg C J. Ten simple rules for getting started with command-line bioinformatics[J]. 2021.
这篇关于学习生物信息学的十条准则的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!